ATTENTION - automatic translation, it is recommended to read the article in the original language
31-year-old boy, normal weight, smoker; operated in 2011 for germinal carcinoma of the left testis treated with orchiectomy and retroperitoneal lymphadenectomy on left paraaortic residue in 2011, with subsequent chemotherapy interrupted due to intolerance. At the beginning of January 2021 he begins to complain of lack of appetite and non-specific asthenia; in February he develops significant bilateral sideerocervical swelling, also involving the base of the neck, clearly visible even on inspection. The blood analyzes show an important anemization with hemoglobin 8.9 gr \ dl.
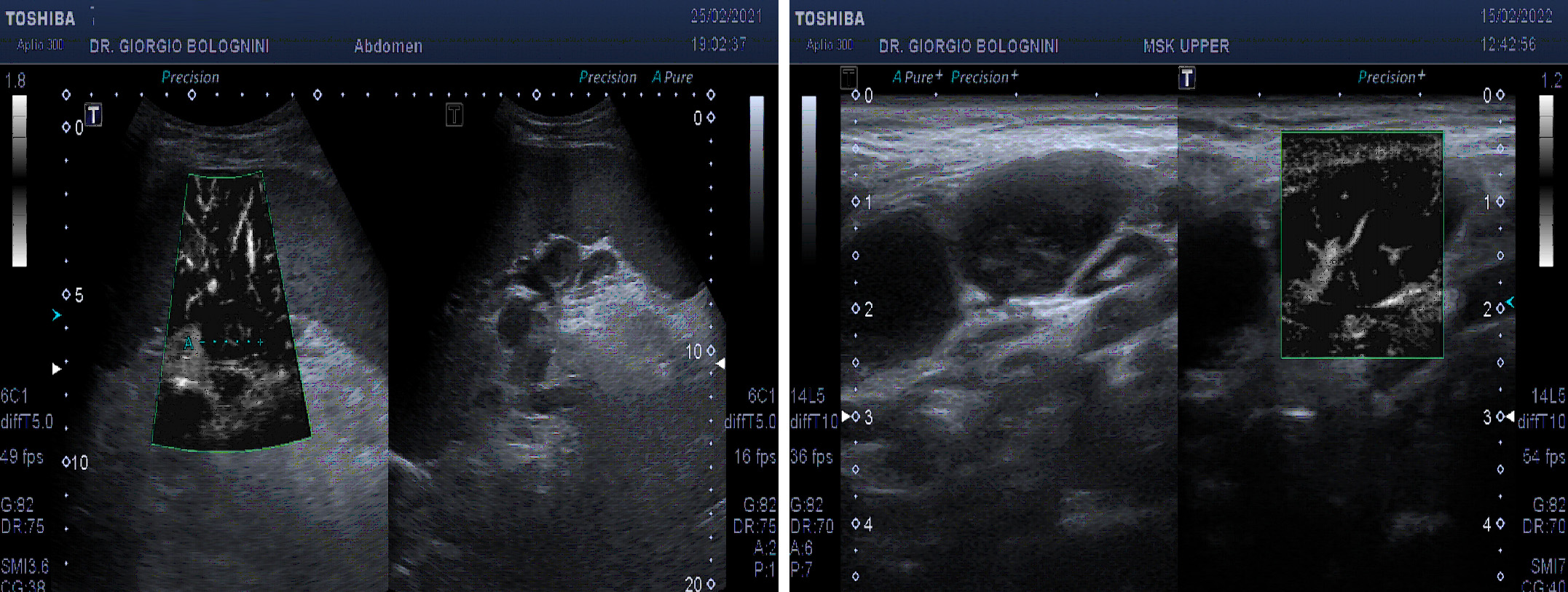
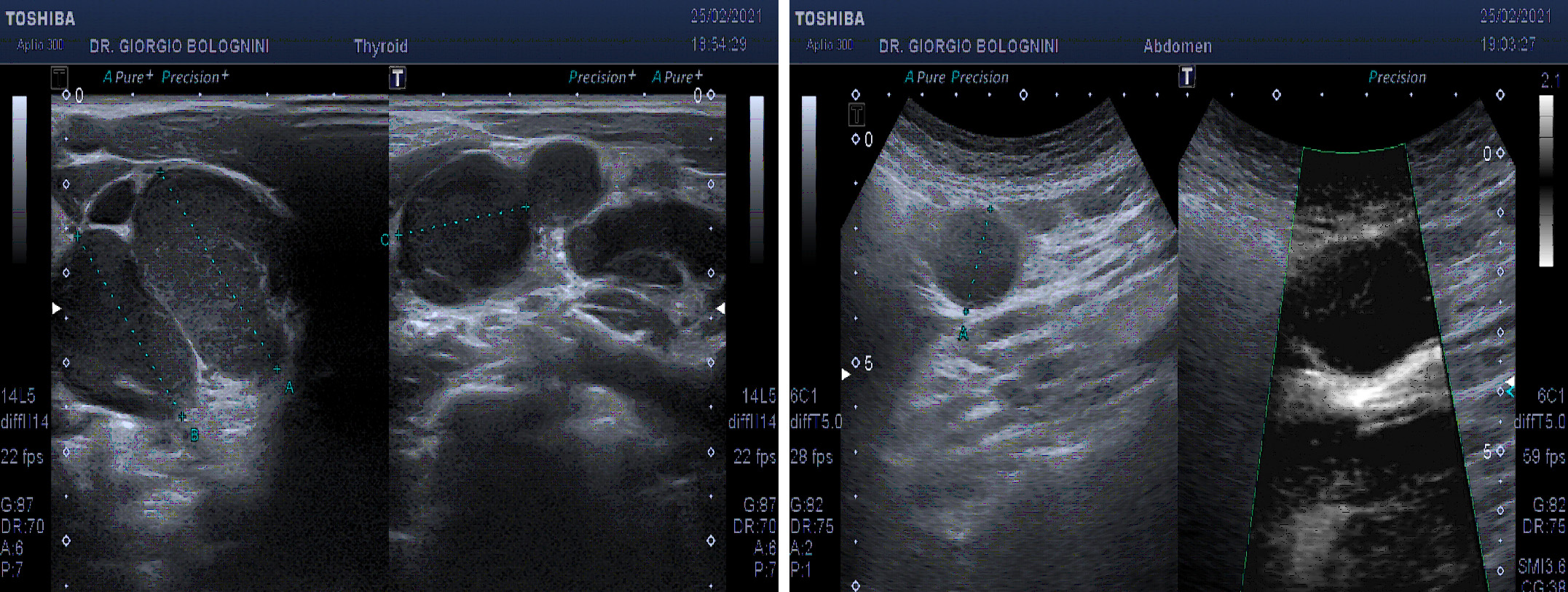
The ultrasound of the superficial lymph node stations showed a picture of ubiquitous laterocervical, supra and subclavicular, bilateral axillary lymphadenomegalies, all with pathological characteristics (disappearance of the hilum, multiple vascular poles, marked hypoechoicity), with only saving of the inguinal lymph node stations. In addition, the examination showed marked splenomegaly (diameter of 22x8cm) with finely granular parenchymal ecostessitura, lymphadenomegaly of a frankly evolutionary type at the splenic hilum (the lymph nodes greater than about 22mm), other pathological lymph nodes at the level of the left iliac axis (at least three glands of maximum size 25mm); multiple small hypoechoic lymphodes in the para aortic and in the caval interport, lymph node with aberrant vascoalrization in the peritoneal fat anterior to the superior mesenteric artery, of about 25mm.
Performs a total body CT scan showing the lymph node package in the mediastinal area of 37mm and in the peripancreatic area with the greater of about 50mm.
He is therefore sent for a haematological examination at the IRCCS in Milan for suspicion of lymphoproliferative disease. He performs a biopsy of the major lymph node in the left lateral cervical region which, however, shows a picture of non-unique interpretation, orienting towards a reactive picture with a rich plasma cell component, in the differential diagnosis between Castleman's disease and an IgC4 related disease. It also performs the BOM which is hypercellulated for age with marked polyclonal plasmacytosis and a slight increase in the fibrotic reticulum (MF1). On blood tests, presence of predominantly polyclonal hypergammaglobulinemia and anemia: GB 6570 (N38%, L 33%, M 10%, Eo 17%), Hb 8.8 mg \ dl, total proteins 13.8 gr \ dl, gammaglobulins 60.9% CM IgG lambda 7 gr \ dl, IgA 359 mg \ dl, IgG 8251 mg \ dl, IgM 24 mg \ dl, IgG4 150 mg \ dl, serum kappa and lambda FLC increased with preserved native FLC, beta2microglobulin 5.38 mg \ dl. Viral serologies for CMV and EBV positive for IgG with IgM negative, IgG anti toxoplasma 95.70 and IgM positive, PCR for EBV, CMV and toxoplasma negative, Quantiferon negative. The histological review at the pathological anatomy of Siena showed "architecture subverted by the widespread proliferation of plasma cell elements, sometimes binucleated, sometimes immature with a plasmablastic aspect (CD20 +, CD38 +, MUM-1 +, KI67 proliferating share of about 50%), with a kappa ratio \ inverted lambda (kappa \ lambda 1: 2), documentable monoclonal rearrangement of immunoglobulin genes on a polyclonal background; the set of findings was therefore compatible with peripheral B-cell lymphoma, type B lymphoma of the marginal area with marked plasmacytic \ plasma cell differentiation, with subsequent evaluation of the MYD88 mutational status to exclude lymphoplasmacytic lymphoma ".
The subsequent review by Prof. Stefano Pileri of the IEO in Milan argued for a HHV8-negative plasma cell Castleman disease (with the clarification that a monoclonal rearrangement of IGH can sometimes be found in this disease) (Soulier J et al, Blood 1995 ). The patient therefore started an infusion therapy with Siltuximab monoclonal antibody, with relative clinical well-being, and carries out the oncological follow-up every six months at my ultrasound clinic.